Original Structure: Magn.
Reson. Chem., 43, 264 (2005)
Revised Structure:
Magn. Reson. Chem., 44, 571 (2006)
This example has been brought to my
attention by Heinz Kolshorn from University of Mainz - both datasets
(and also both articles) under discussion here, are not within my
internal CSEARCH-environment as per March 28th, 2008!
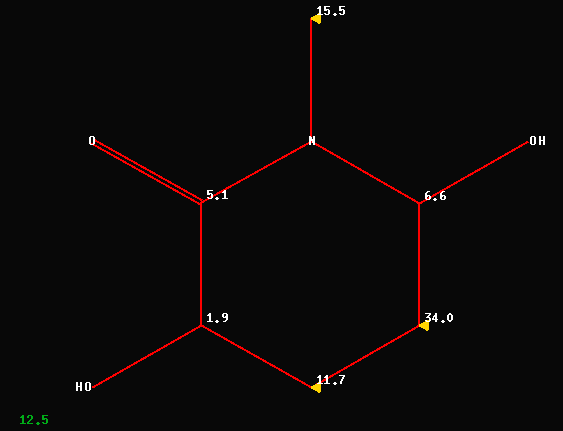
The following C-NMR data (172.9/S,
70.1/D, 69.6/D, 62.7/T, 43.2/Q and 38.3/T) have been used to elucidate
compound #1 (numbering scheme from MRC-2006) as
3,6-dihydroxy-N-methyl-2-piperidone. The investment of approximately
10msec of CPU-time reveals an average deviation of 12.5ppm between
experimental CNMR-data and the predicted shift values as shown below. 3
Carbons (out of 6!) have deviations between 11 and 34ppm !
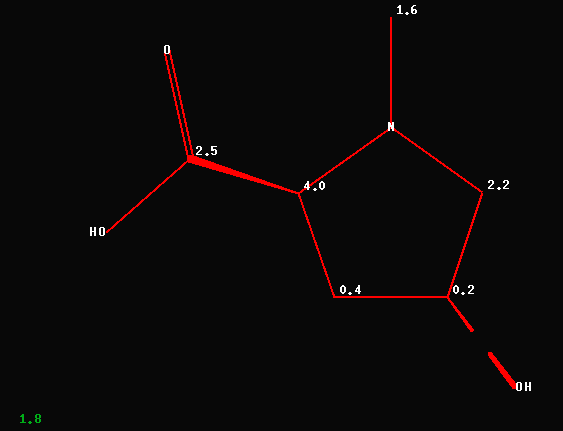
The revised structure by T. Winkler
leads to a known proline-derivative; the average deviation between
experimental and predicted carbon chemical shift data is now 1.8ppm as
shown below.
All predictions have been done using
CSEARCH-NN-technology ommitting solvent-dependant prediction. Compound
#1 has been predicted without stereochemical information, whereas the
prediction of compound #3 was done using stereochemical information.
Both
structures (and also both articles) are not avaialable within all the
data at my internal CSEARCH-installation !
This
example leads directly to the central problem of our publishing
procedures:
The
facts are:
- The first structure proposal is far away from being compatible
with the C-NMR data
- The peer-reviewing process was unable to detect the problem
- An automatic reviewing process ('robot-referee') using
CSEARCH-Technology can easily detect this (dramatic) incompatibility
Conclusion:
Apply
spectrum prediction whenever possible
- Daily routine during generation
and interpretation of NMR-data
- The right time is exactly when
you see your spectra for the VERY
FIRST TIME!
- Check again during preparation
of a manuscript
- Check again during the
peer-reviewing process ('robot-referee')
Page
written by: Wolfgang.Robien(-at-)c13nmr.at
Page written on: March
28th, 2008